ChromImpute和ChromHMM一样,同样是来自Ernst and Kellis 的作品。
背景:
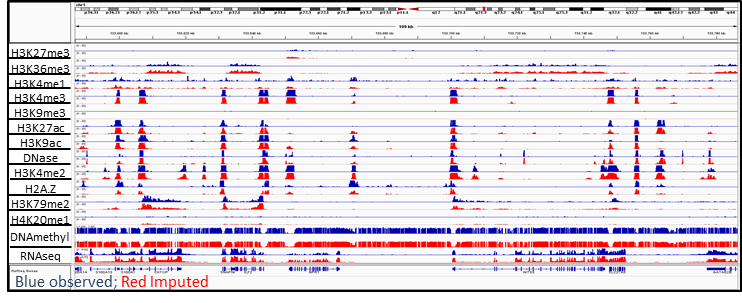
由于样本可得性问题,导致目前公开的表观基因组数据集中大量样本或者大量染色质标记的数据存在缺失。这样的数据会导致不同样本比较时或者不同染色质标记比较时,存在数据不匹配问题。因此ChromImpute被开发出来用于对染色质标记-样本面板中缺失数据进行推断和填充。收集的样本为111 Roadmap数据集和 16 ENCODE数据集,涉及染色质标记包括组蛋白修饰印记、DNA可及性数据、DNA甲基化数据、RNA-seq数据,以及其它信号坐标数据等。
一、数据预处理
- 来自于Roadmap的组蛋白修饰和DNase信号数据:拟定序列富集的信号背景为泊松分布,对应的信号强度表示为 -log10Penrichment;
- 对于RNA-seq数据,将均一化处理的表达量信号标准化为RPKM值(reads per kilobases of exon model per millions reads),用于去除长度和样本大小的影响,标准化的 RPKM 值是基于将未标准化的信号值乘以 10 9,然后除以读取长度和外显子读取数的乘积来计算的,不包括线粒体、核糖体和信号值的前 0.5%。最终信号值表示为 log2(RPKM+1) 。
- 然后用overlapping的25bp-bin内的平均信号值处理方式,将组蛋白修饰信号、DNase信号和RNA-seq信号数据转换为25-bp精度的信号值。
- 对于DNA甲基化数据,我们使用均一化处理的全基因组亚硫酸氢盐测序(Bisulfite-seq)数据,它提供了所有 CpG (至少有3条reads覆盖)中每个碱基的甲基化比例。对于CpG甲基化缺失值,用全基因组DNA甲基化水平的平均值进行填充。
二、ChromImpute算法原理
预测指定样本的指定mark的信号值,依赖于两类特征:
- 同一样本中其它mark的信号值;
- 同一mark其它样本的信号值。
除了目标样本,其它有可利用的mark信号数据的样本,整合它们这两类特征,训练预测器。然后用训练好的预测器集合对目标样本的信号值进行预测,最后取所有预测器的预测平均值作为集成预测器的输出。集成预测可以平均化所有预测器的偏差。
具体内容:
令Oc,m,p为样本c在位置p处,mark m的观测信号值;令Mc,m为用于预测mark m 作为训练集的样本c中的mark集合;令Cm为在mark m 处存在信号值的样本集合;令mt为目标mark,ct为目标样本。对于样本 ct' ∈ Cmt{ct},分别定义特征,来预测样本 ct 中 mark mt 的信号值。对于一个训练样本 ct',令MI 为 Mct,mt ∩ Mct',mt \ {mt},表示靶标样本ct和训练样本ct'中可以用来预测mark mt' 的信号值的共有mark数据集,然后定义两类特征来预测样本 ct'在基因组目标位置p处 mark mt的信号值。
- 基于映射在同一样本中的其他标记集的特征。定义每个标记m ∈ MI的特征集 Sm,n,其中 n = 500 i或n = 25 i (int i = -20,...,20 )。特征Sm,n赋值为 Oct', mml , p + n。
p +n是指在相同的染色体上的位置p,上下游范围 n 的区域。研究中对应于在目标位置 500 bp 内每 25 bp 的特征值以及目标位置上游和下游 10,000 bp 内每 500 bp 的特征值。 - 基于其他样本中目标标记的特征。定义每个标记m ∈ MI的特征集 fm,g,k , 其中g ∈ { local,global }, k = 1,...,min(10,| CI |),而CI 定义为Cmt ∩ Cm \ { ct′ ,ct }。CI对应于具有目标标记的所有样本,并且这些标记将被用于评估样本间的相似性,不包括总体目标样本和用于训练预测器的样本。fm,g,k 值为 Oc,m,p,其中cj是CI中,按dm,g(ct′, c) (c ∈ CI)的递增值排序时,处于排序位置j 的样本。如果g = global,则 dm,g(ct′, c) = 1-ρ(Oct', m Oc,m) ,其中ρ是在样本ct'和c中全基因组范围内mark m信号值的 Pearson 相关系数 。如果g = local,则在位置p处,采用目标mark附近500bp的信号值,dm,g(ct′, c) = (Oct', m,p+25i Oc,m,p+25i)2
上面定义的特征集 Sm,n和fm,g,k 用来构建特征向量。特征向量用于基于样本ct'预测样本ct的信号值。
预测器采用回归树
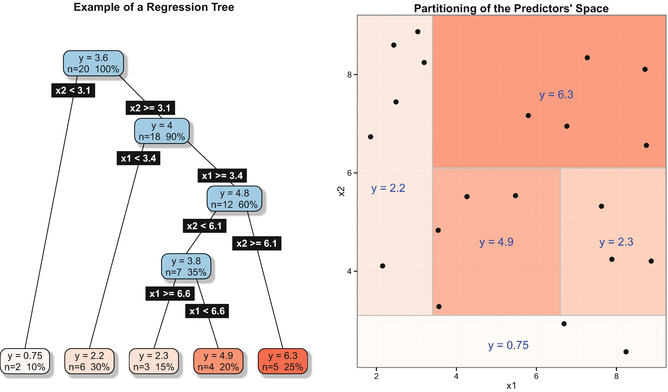
回归树指用决策树方法做回归任务。分析的数据类型是连续性信号数据,即不同表观遗传标记的信号强度值,而用来进行树构建的特征集为上一步处理得到的两组特征:基于映射在同一样本中的其他标记集的特征和基于其他样本中目标标记的特征。
构建的回归树T的描述:Split nodes集合S(隐藏层,样本集合进一步聚类形成的内部结构),Leaf nodes集合N(可以理解为样本)。
定义Split nodes s∈S,可用一个四元组(f,v,l,r)表示,
参考文献:
Ernst, J., Kellis, M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 33, 364–376 (2015).