从bam文件中提取fastq reads
方式1:Bedtools
if single-end reads:
bedtools bamtofastq -i input.bam -fq output.fastq
if pair-end reads:
samtools sort -n input.bam -o input_sorted.bam (sort reads by identifier-name (-n) )
bedtools bamtofastq -i input_sorted.bam -fq output_r1.fastq -fq2 output_r2.fastq
存在的问题:
如下图,从上至下依次是read1,read2,read3,read4。有两种序列。其中read1和read2同一条read mapping到基因组不同区域后保留下来的重复reads,read3和read4同理。

以下是利用bedtools工具提取出来的fastq文件。对bam文件reads name集合取unique后,发现unique reads的数目和bedtools提取的reads数目一致,而且从bedtools fastq结果文件可以看到,bedtools并没有在有重复reads的bam文件中正确区分fastq1和fastq2中的对应paired reads。而且可以推断出它的做法是:优先去主染色体上的重复reads或随机选取一条重复reads保留,如果保留下来的是负链上的reads,则反向互补转换后保存下来,然后拷贝至两个fastq文件中。
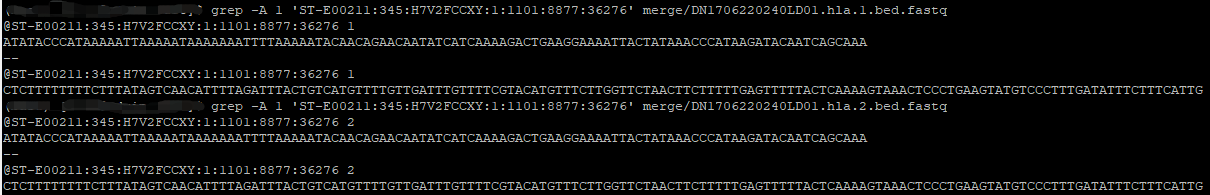
再看一组reads,这组虽然重名,但是有三种序列,如下:

read1和read2可以看出来是同一条read mapping到基因组不同区域后保留下来的重复reads,而read3和read4序列末端是一致的,说明其中短的可能由于区域选择问题被截断,而实际上这两条也是重复reads。bedtools bam2fastq后,
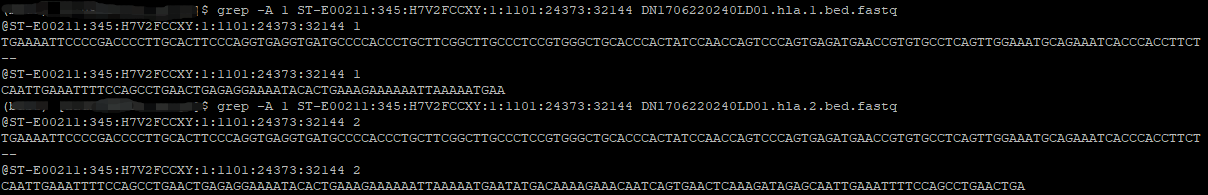
可以看到,bedtools的做法和之前一样,从read1和read2中选择一条拷贝至两个fastq文件,从read3和read4中选择一条拷贝至两个fastq文件。
bedtools在带有重复reads的bam文件中的这种bam2fastq方式明显是存在问题的,实际上bam文件中FLAG值中保留着reads的paired信息,可是bedtools却没有识别到。解决方式一是去除重复后再进行bam2fastq(也不晓得可不可行),二是用Picard或者samtools来进行bam2fastq。
方式2:Picard
java -Xmx2g -jar SamToFastq.jar I=SAMPLE.bam F=SAMPLE_r1.fastq F2=SAMPLE_r2.fastq FU=SAMPLE_unpaired.fastq
F2 —— to get two files for paired-end reads if reads are paired (R1 and R2)
FU—— to keep unpaired reads
-Xmx2g——allows a maximum use of 2GB memory for the JVM
存在的问题:
截取基因组局部区域的bam文件提取fastq reads,由于大量之前被FLAG标记为paired-end的reads被截断后,失去了其中一条,结果bam2fastq时找不到另一条了,Picard会报错终止。
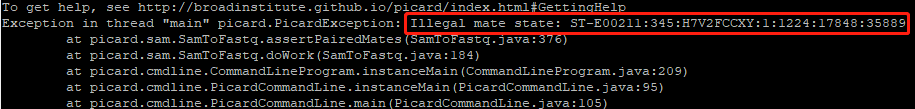
解决方式:用samtools来进行bam2fastq。
方式3:Samtools
samtools sort -n SAMPLE.bam -o SAMPLE_sorted.bam (sort paired read alignment .bam file (sort by name -n))
samtools fastq -@ 6 SAMPLE_sorted.bam -1 SAMPLE_R1.fastq -2 SAMPLE_R2.fastq
samtools bam2fastq 目前看起来没什么毛病。
Reference:
- https://www.metagenomics.wiki/tools/samtools/converting-bam-to-fastq
- https://www.jianshu.com/p/901c1cdf8a45