HLA-VBSeq算法
一种可在8-digit水平对WGS数据进行HLA分型的方法。其基本流程如下:
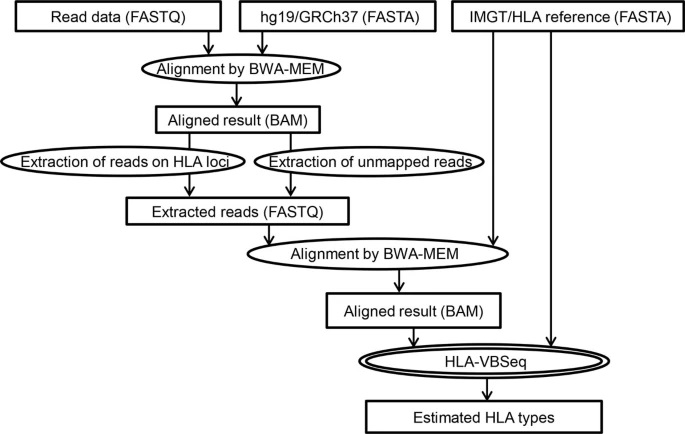
模型构建:
HLA-VBSeq的算法核心,就是将HLA区域或者可能来自HLA区域的reads(包括由于多态性过高而无法mapping到参考基因组的reads)在IMGT/HLA数据库中找到最匹配的单体型,从而对reads分型。HLA-VBSeq采用的是生成式模型,如下图:
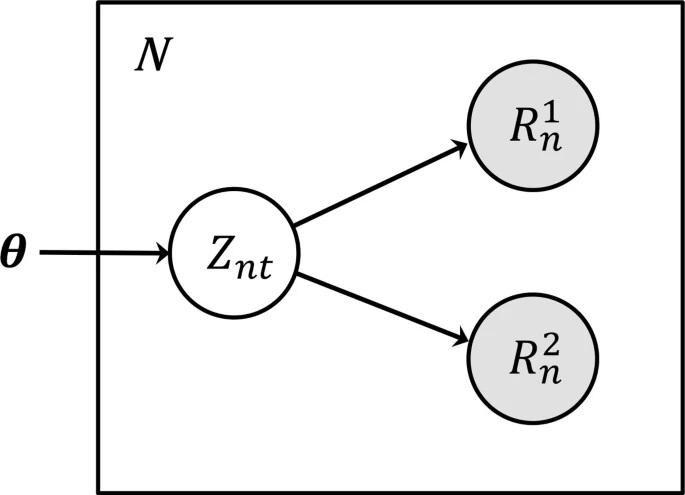
表示reads丰度(基于等位基因长度标准化后的reads覆盖深度)
对于paired-end reads,两条reads分别为,。是示性变量,该read n匹配到参考HLA等位基因t上,则为1,否则为0,即:
这样,贝叶斯模型可定义为:,即已知reads可以匹配到对应HLA等位基因上,那么reads的丰度分布应该是怎样。
贝叶斯分布的先验分布常用Dirichlet分布表示,有
其中,是常量,,T是考虑的HLA等位基因数目,而是控制模型复杂度的超参。该方法中,使用观测数据的最大对数边缘似然值作为该模型的统一超参
变分贝叶斯推断
由于算法需要根据观测数据估计reads丰度的后验分布,而模型中存在隐变量,导致模型难以求解,因此采用变分贝叶斯推断(Variational Bayesian Inference)的方法对求近似解(可参考EM(期望最大化)算法)。
E Step:
对于read n,其对于HLA等位基因t的期望read count数,可由当前估计的read丰度参数计算得到:
M Step:
read在每个HLA等位基因上的丰度可由估计的期望read count数计算得到:
其中
通过E-M迭代,直到满足收敛条件。
通过确定相对于HLA等位基因的最优read alignment,确定read所对应的HLA单体型
在上述算法收敛后,则可通过计算每条read在HLA参考等位基因中匹配的read count数(观测值)相对于每个HLA参考等位基因的期望read count数,是否显著符合,从而判断该read对应哪种HLA单体型。
由于存在测序误差(相对于参考HLA序列的替换、删除和插入)和比对误差,设置了 HLA 等位基因覆盖深度的阈值。在我们的分析中,我们将阈值设置为覆盖深度的 20%(即,如果数据平均为 30x,那么我们使用 6x 作为阈值)。对于每个 HLA 基因座,二倍型确定如下:
- 如果没有超过阈值的等位基因,则认为没有足够的reads 来识别正确的HLA 类型,因此这些reads无法分型。
- 如果只有一个等位基因通过阈值,且覆盖深度大于或等于阈值的两倍,则认为reads所在的HLA 基因座是该HLA 等位基因的纯合子。如果覆盖深度小于阈值的两倍,则称该基因座的等位基因为杂合的。
- 如果有两个或更多等位基因通过阈值,则根据覆盖深度从高到低对等位基因进行排序。匹配度最高的前两个等位基因被选为 HLA 类型的候选者。如果最匹配的HLA等位基因的reads覆盖深度是第二匹配的两倍以上,则 reads所在的HLA 基因座被设定为最匹配等位基因的纯合子。否则,HLA 基因座被设定为最匹配和次匹配等位基因的杂合基因型。
References:
- Nariai, N., Kojima, K., Saito, S. et al. HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genomics 16, S7 (2015).