HLA-HD算法
HLA-HD是目前HLA基因分型Availablity 和 Accuracy兼顾的一款软件,分辨率可达到3-field,即6位分型精度。
算法细节:
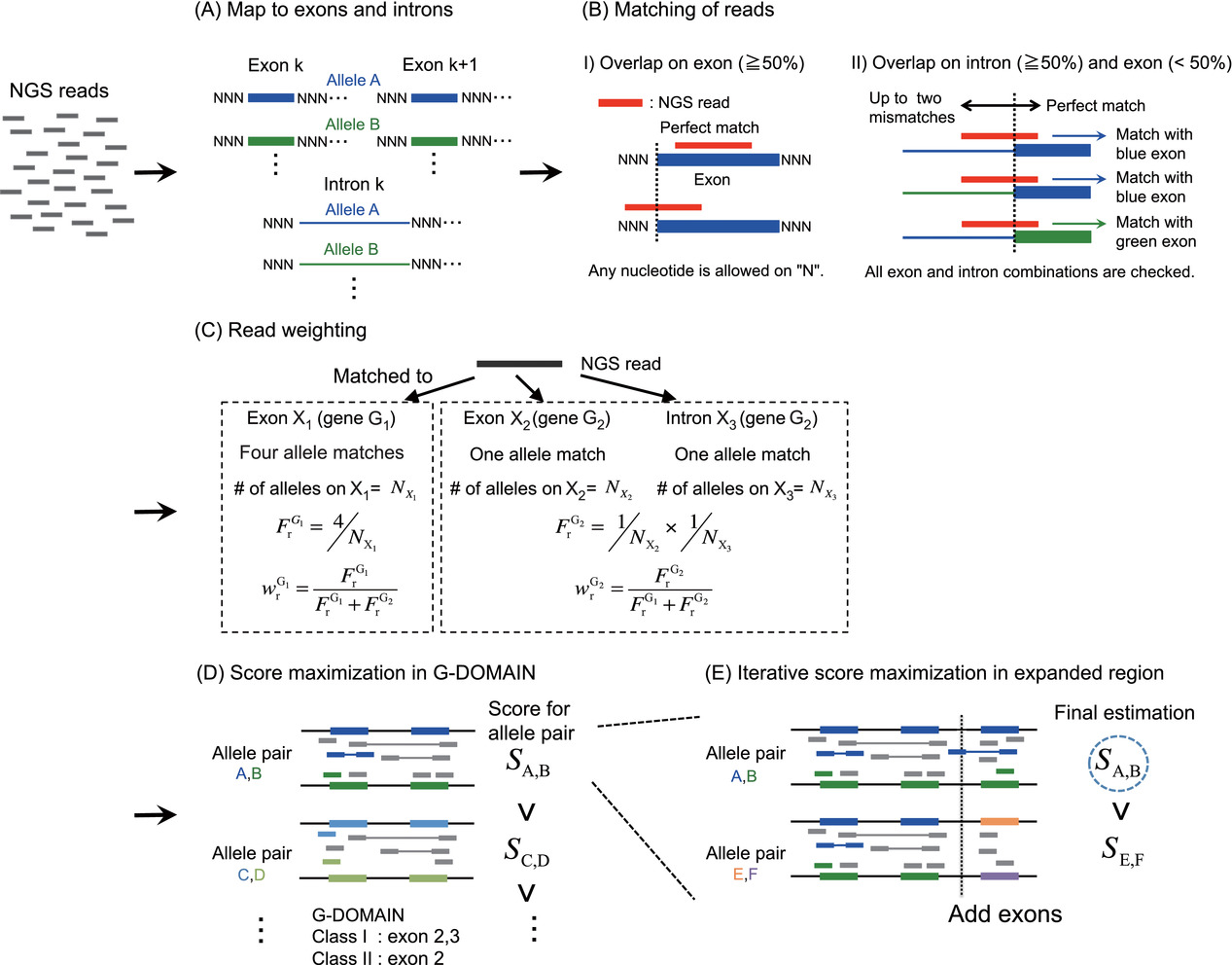
第一步,HLA字典的构建:
下载IPD-IMGT/HLA数据库收录的HLA等位基因字典(HLA dictionary,该字典包含关于33个HLA基因的10684个等位基因,release 3.15.0,随着版本不断更新,等位基因的数目也会不断更新)。将HLA等位基因序列分解为外显子(exons)单元和内含子(introns)单元,并在每条序列两端加上150个“N”(因为NGS reads的最大长度大约是300bp左右,因此在reference HLA等位基因两端加上150个“N”核苷酸作为参考序列)。
第二步:序列比对:
将NGS reads比对到构建好的HLA字典对应的不同等位基因序列(exons和introns序列)上。reference HLA等位基因两端的“N”不贡献比对分数,不影响比对分数。
若是paired-end测序,需要将forward-end和reverse-end的reads分别比对到HLA字典中。若是single-end测序,则直接比对到HLA字典中。
依赖软件参数:
Bowtie2 --n-ceil L,0,0.5 --score-min L,-1.0,0 --a
--n-ceil L,0,0.5:该参数用来设置reads比对的最大模糊匹配碱基数。
以函数f(x) = a + b * x来计算,x为read长度。
L,0,0.5表示a=0, b=0.5,若read长度为150bp,则最大模糊匹配数为75bp。
--score-min L,-1.0,0:该参数用来设置reads比对的最小有效比对分数。
同一样以一个函数f(x) = a + b * x来计算,x为read长度。
L,-1.0,0表示a=-1.0, b=0,若read长度为150bp,则最小有效比对分数为-1。
--a:当get到一个质量分数合格的有效alignment时,继续搜索该read与其它基因组位置或HLA等位基因的有效alignment。通过穷尽搜索得到比对分数最高的alignment。若最后得到的高质量比对的alignments存在两个或多个,则从中随机选择一个最为输出alignment。
第三步:alignments过滤:
上一步构建的alignments仅保留符合以下要求的:
- 与reference中外显子序列完全匹配,没有错配
- 与reference中内含子序列最多存在两个错配
- 当reads有超过50%的长度在reference中外显子序列(完全匹配)中,
将该reads分配给外显子序列 - 若reads有超过50%的长度在reference中内含子序列(允许两个错配)中,
将该reads分配给内含子序列 - 如果一条"best match"到reference中外显子序列的read,
那么不管该read是否存在内含子序列,都不会对该read是否能分配到到外显子序列造成影响; - 而如果一条best match"到reference中内含子序列的read,
那么该read内含子邻近区域如果存在外显子序列,则需要留待进一步分析(见图B)。
第四步:计算比对权重分数
因为一条read可能比对到不同基因外显子或内含子区域,因此需要计算比对的权重分数,进行区分判定。
为了计算一对等位基因比对的得分,将按照图C那样,对匹配到外显子区域或内含子区域的reads根据匹配的等位基因数目赋予对应的权重。
令为read 匹配到外显子或内含子上匹配的等位基因数目;
令为HLA字典中含有该外显子或内含子区域X的等位基因数目。
则
- 对于一条single-end read,令read 和基因的权重为,则有
其中表示所有HLA基因的集合;
其中表示
反映了该read r在该基因区域的匹配分数占所有匹配基因区域的匹配分数的权重,体现了该read在不同匹配区域的适配程度。
反映的是该read r匹配到该基因G各外显子和内含子区域的可能性(匹配的等位基因数目占该位置所有等位基因数目的比例)之积。
在公式(2)中,
表示基因所有外显子和内含子的集合
- 对于一对由正链read 和负链read 构成的paired-end reads,其权重为
第五步:计算reads最适配的等位基因类型
令为匹配到基因G外显子X等位基因A处的reads集合,包含匹配到内含子区域但与外显子区域重叠的reads,并且正链reads和负链reads各自单独计算。
目标外显子区域等位基因A的权重分数定义为:
其中T表示所有外显子集合,表示read r与外显子X重叠的长度
对于paired-end reads,用替换即可。
一开始时,T设定为G-domain(抗原结合沟)对应的外显子区域(exons 2 and 3 of HLA-A, HLA-B, and HLA-C or exon 2 of HLA-DRB1, HLA-DQB1, and HLA-DQA1)。
将简化为
类似于Single-end reads,同时匹配到等位基因A外显子区域的paired-end reads的比对分数定义为:
其中表示和在匹配区域的overlap长度,然后可知表示paired-end两条readsp匹配到该外显子区域的总长度。
对于Single-end reads,
因为人是二倍体生物,因此以下将要找到该区域所有reads能匹配到的最适的两个HLA单体型。
接下来,令 为同时匹配到等位基因A和B上的reads集合(paired-end reads),表示匹配到等位基因A,而没有匹配到等位基因B上的reads集合(paired-end reads)。对于这些reads集合的匹配分数可以这么计算:
选择具有最大匹配分值的等位基因对作为适配等位基因类型。
当与的比值过高或过低时,可以认为该HLA基因是纯合的,只有一个最适匹配HLA单体型。有两种计算等位基因是否是纯合的方式:
当 或者 时,可以认为该等位基因A是纯合的。
第六步:增加分型精度
到目前,对read进行分型,采用的参考HLA基因型仍是在G-domain水平。当在G-domain水平无法对reads进行分型时,则需要增加HLA字典中G-domain以外的其它外显子区域的等位基因到T中,以增加分型精度(见图E)。这样,则可以突破G-domain分型精度,达到3-field(6-digit)分型精度水平。
Reference:
- Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA-HD: An accurate HLA typing algorithm for next-generation sequencing data. Hum Mutat. 2017;38(7):788-797. doi:10.1002/humu.23230
- https://www.genome.med.kyoto-u.ac.jp/HLA-HD/