题目: The evolutionary history of 2,658 cancers
单位:PCAWG Consortium
期刊:Nature
发表日期:2020-02-05
摘要
Cancer develops through a process of somatic evolution1,2. Sequencing data from a single biopsy represent a snapshot of this process that can reveal the timing of specific genomic aberrations and the changing influence of mutational processes3. Here, by whole-genome sequencing analysis of 2,658 cancers as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The
Cancer Genome Atlas (TCGA)4, we reconstruct the life history and evolution of mutational processes and driver mutation sequences of 38 types of cancer. Early oncogenesis is characterized by mutations in a constrained set of driver genes, and specific copy number gains, such as trisomy 7 in glioblastoma and isochromosome 17q in medulloblastoma. The mutational spectrum changes significantly throughout tumour evolution in 40% of samples. A nearly fourfold diversification of driver genes and increased genomic instability are features of later stages. Copy number alterations often occur in mitotic crises, and lead to simultaneous gains of chromosomal segments. Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer, and highlight opportunities for early cancer detection.
研究背景:
类似于物种进化,体细胞群体中也普遍存在突变和选择。大部分突变是中性的,受连锁效应影响作为“乘客”受到“选择”作用;剩下的突变大多也是有害的,在负选择过程中慢慢被清除;只有少量突变是对细胞生存是有利的。目前对于正常体细胞进化和癌细胞进化突变出现时序谱之间的差异了解非常少。因此,本文利用 PCAWG 数据集来表征来自 38 种癌症类型的 2,658 名独特捐赠者的 2,778 份癌症样本的进化历史。作者推断染色体进化的时间和模式,并学习每种癌症类型样本的典型突变序列。然后,作者定义了肿瘤进化的广泛时期,并研究了驱动因素和突变特征在这些时期之间如何变化。使用类似时钟的突变过程,作者将突变时间估计映射到近似实时。总的来说,这些分析勾勒出了癌症的典型进化轨迹,并相对于诊断点实时绘制它们。
方法部分:
一、拷贝数增加时间估计
- 突变等位基因频率(VAF)的期望值 E[X] = nmfρ/[N (1 − ρ) + Cρ]
X:reads数目;n:位点的reads覆盖度;m:携带该突变的等位基因数目;
f:携带给定突变的clone数目 (f = 1 for clonal mutations);
N:标准拷贝数 (2 on autosomes, 1 or 2 for chromosome X and 0 or 1 for chromosome Y);
C:肿瘤的总拷贝数;ρ:样本纯度 - 在等位基因拷贝数m下突变数目为nm下,可以推断拷贝扩增的时间:
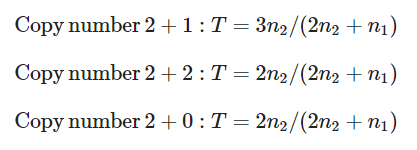
2+1对应主等位基因和次等位基因拷贝数分别为2和1。没有考虑2+2这种情形是因为两个等位基因同时发生拷贝数变异的概率很低。
二、突变时间追溯
一些概念的区分:
根据点突变覆盖的reads数目可在拷贝事件发生情况下,将变异区分为早期克隆(early clonal),晚期克隆(late clonal),在拷贝事件没有发生的情况下,将变异区分为克隆(clonal)和亚克隆(subclonal)。见Fig1.a
- Early clonal:mutation on >=2 copies per cell (f=1 and m>1)
- Late clonal: mutation on 1 copy per cell, no retained allele (f=1, m=1 and C>2)
- Clonal: mutation on 1 copy per cell, either on amplified or retained allele (f=1)
- Subclonal: mutation on < 1 copy per cell (f < 1)
三、驱动突变时间追溯
计算PCAWG提供的癌症驱动点突变,在四个时期中,early/late clonal的odds-ratios和clonal(包括early,late和unspecified clonal)/subclonal的odds-ratio。非驱动区域具有相同拷贝数组成的片段上的突变用于计算背景分布,1000 bootstraps。
四、整体时间推断
对于每对驱动点突变和往复拷贝数变化,确定其早期、晚期和未指定状态。
五、突变印记时间推断
三核苷酸替换突变印记,在将点突变分配到四个不同时期后,拟合样本中观测印记活性。根据多项式分布用似然比检验检验突变图谱和时间点的差异。同时利用非负线性最小方差方法计算不同时间的暴露因素影响。
六、WGD(全基因组复制)和MRCA(最近共同祖先)的实时估计
- 对基因组中NpCpG CpG>TpG突变进行计数。对于皮肤癌样本中CpCpG>TpCpG突变不予考虑,因为和UV诱导的突变重叠。考虑基因组大小影响,将突变数目标准化到有效基因组大小中,定义为1/mean(mi/Ci)。mi是个突变的等位基因拷贝的估计数量,Ci是该位点的总拷贝数,从而标准化最终拷贝数和变化时间。
- 分层贝叶斯线性回归适合将诊断年龄与按比例缩放的突变数量相关联,通过跨癌症类型的共享伽马分布确保正斜率和截距。对于具有多个时间点的肿瘤,计算了诊断和复发之间共享的一组突变 ( nD ) 以及那些特定于复发的突变( nR )。速率加速度计算为:a = nR / nD × tD / tR。该分析是针对所有替换突变和 CpG>TpG 突变分别进行的。
- 根据这些分析,选择的大多数癌症类型最终拷贝数增加了 5 倍,脑癌较低为 2.5 倍,卵巢癌的值为 7.5 倍。将突变时间中拷贝数增益的估计转换为时间顺序的校正不仅取决于速率加速,还取决于该加速发生的时间。由于这通常是未知的,我们在诊断前对跨越 15 年时间间隔的速率加速进行了蒙特卡罗模拟,大约相当于 60 岁时诊断时间的 25%,注意到在此期间速率增加 5 倍会产生大约 33% 的突变偏移,与我们的数据兼容。假设亚克隆突变发生是全速进行的。亚克隆突变的比例除以鉴定的亚克隆数量,因此保守地假设分支进化。
七、癌症时间表
每种不同时间分析的结果都结合在每种肿瘤类型的癌症演变时间线中。每个时间线都从受精卵开始,一直延伸到每个队列中的中位诊断年龄。WGD 和 MRCA 的实时估计作为锚点,使我们能够粗略地将四个广泛定义的时间段(早期克隆、中期、晚期克隆和亚克隆)映射到患者生命周期中的时间顺序。可以根据联合模型分析中的顺序,将特定的驱动突变或拷贝数改变放置在每个时间范围内。如果它们通常随时间变化(平均变化的 95% 置信区间不与 0 重叠),并且它们非常活跃(对一个时间点贡献至少 10% 的突变),则会显示特征。印记显示在时间线上他们最活跃的时期。
结果部分
一、重建肿瘤生命历程
- 初始时,每个点突变发生在单个细胞的单个染色体上,从而产生具有相同突变的细胞谱系。如果一条染色体基因座发生拷贝数变异,则该等位基因在拷贝前的任何点突变都会出现在两个等位基因拷贝(
一个母体,一个子体)上。这和不依赖于拷贝事件,仅继承于等位基因拷贝或者其它未发生拷贝事件的等位基因座上的突变不同。等位基因拷贝数可以从测序数据中获得,因此可以定义拷贝事件发生前后基因座上的突变,未拷贝的基因座突变。 - 拷贝事件内,克隆突变和非克隆突变的比率可用于估计在克隆进化过程中发生基因座拷贝变异事件的时间点,也可理解为测定相对于拷贝事件,发生在拷贝单元内的新突变的发生时间。
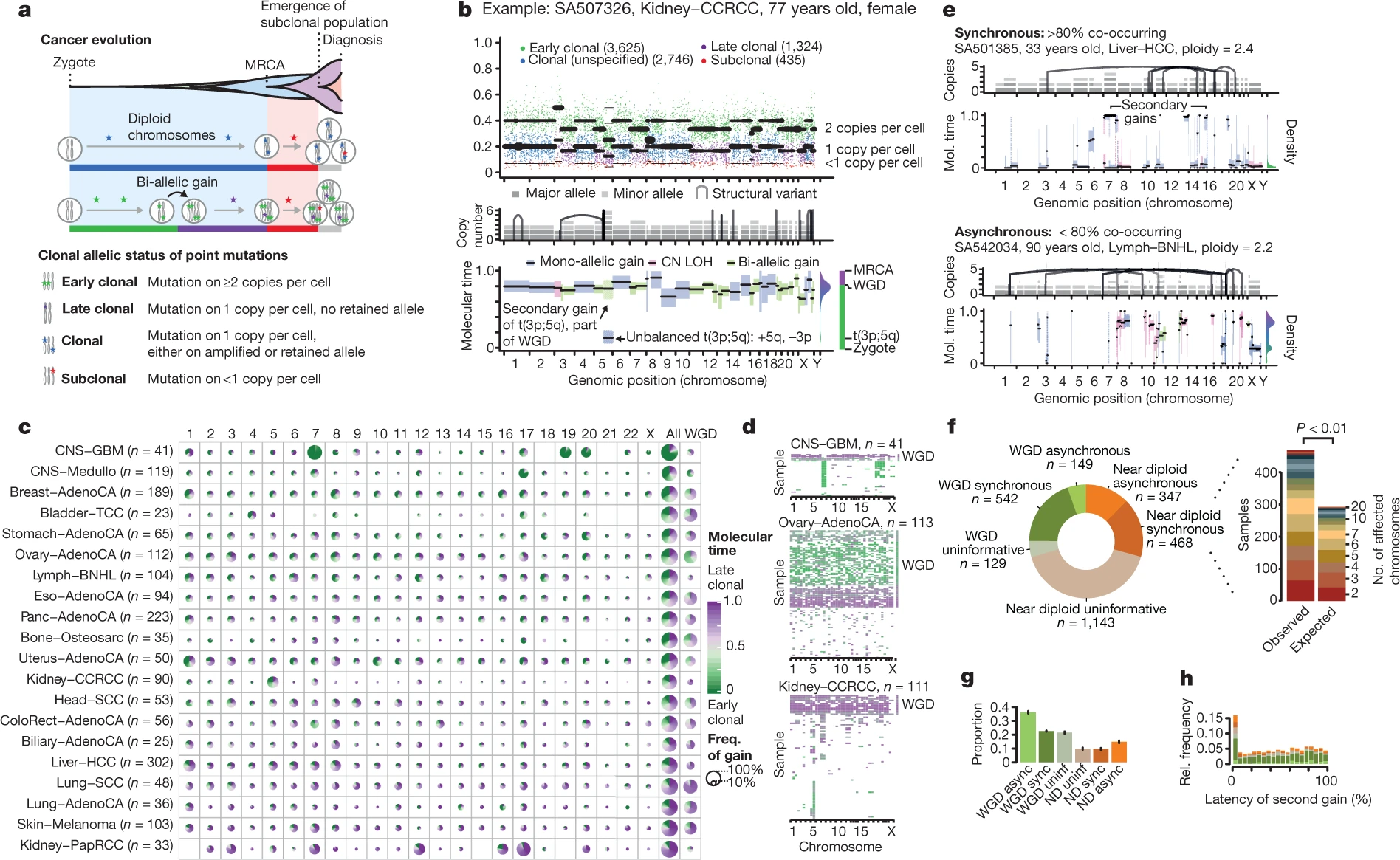
二、拷贝数增加的时序模式
- 染色体突变获得时序谱发生在很宽的分子时间范围内,不同肿瘤类型间存在系统偏差,而同种肿瘤类型不同染色体间也会表现出相似的突变时序分布。在胶质母细胞瘤和髓母细胞瘤中,大部分增益发生在分子时间的早期。相比之下,在肺癌、黑色素瘤和乳头状肾癌中,增益出现在分子时间尺度的末尾。大多数肿瘤类型,包括乳腺癌、卵巢癌和结直肠癌,都显示出相对广泛的染色体不稳定时期,这表明样本中的增益时间非常可变。
- 某些肿瘤类型始终具有特定染色体区域的早期或晚期增益。最明显的是胶质母细胞瘤,其中 90% 的肿瘤包含 7、19 或 20 号染色体的单拷贝增益。
- 同一肿瘤中的拷贝后的突变通常似乎发生在相似的分子时间,指向涉及大多数增益片段的拷贝数增益的间断爆发。
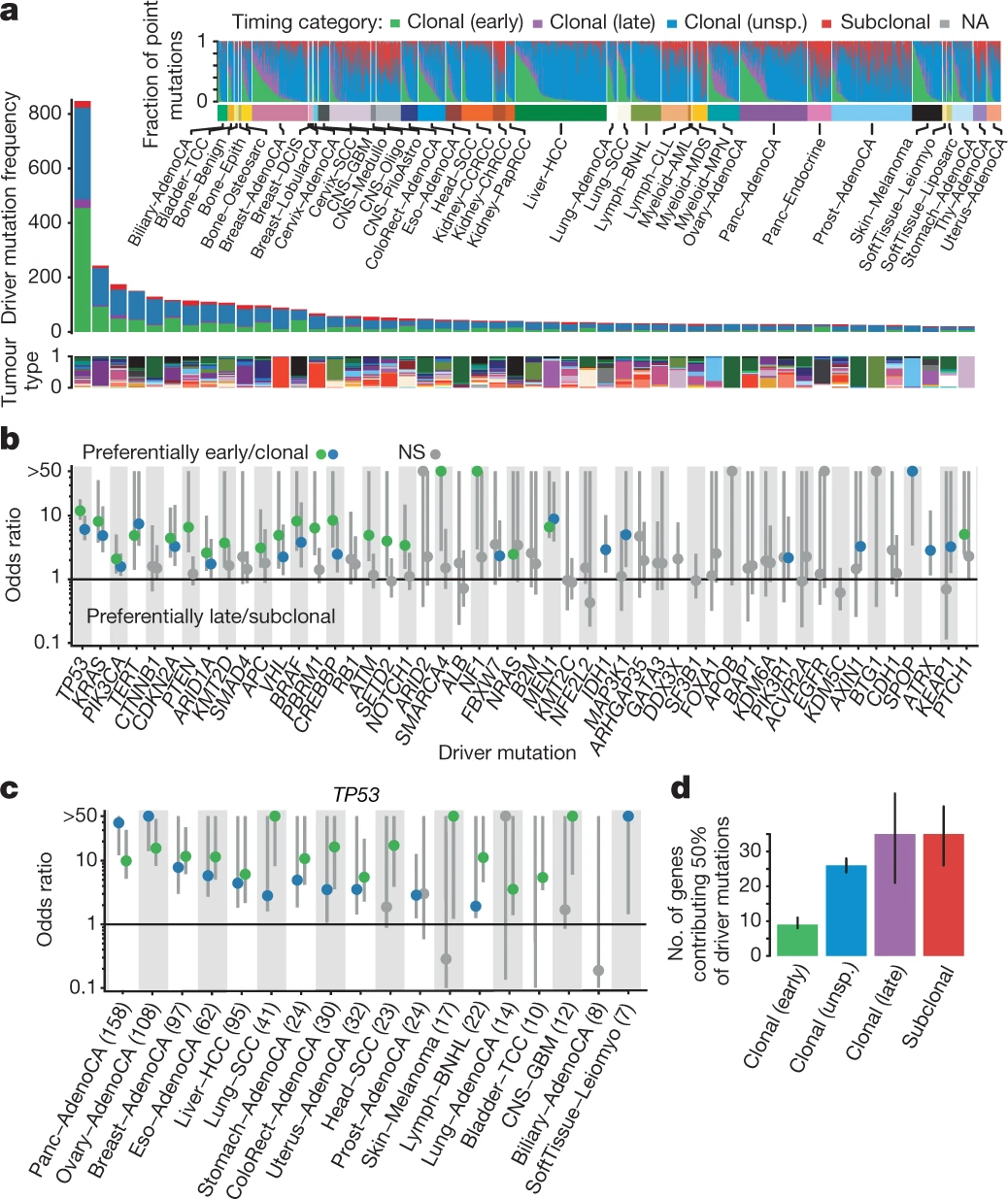
三、肿瘤驱动基因中点突变的发生时间
- 根据前面对体细胞突变的定性分期,癌细胞驱动基因上的突变同理可以进行分期。在 2,583 个样本的 4700 万个点突变中,22% 是早期克隆,7% 是晚期克隆,53% 是未指定克隆,17% 是亚克隆。而在一组 453 个癌症驱动基因中,鉴定出 5913 个致癌点突变,其中 29% 为早期克隆,5% 为晚期克隆,56% 为未指定克隆,8% 为亚克隆。结果表明癌症驱动基因突变在早期克隆和未指定克隆中富集。
- 按照时间推移,所有癌细胞驱动基因点突变多样性在肿瘤后期增加:所有早期克隆驱动突变的 50% 仅发生在 9 个基因中,而 50% 的晚期和亚克隆突变发生在每个大约有 35 个不同的基因,增加了近四倍。结果表明早期癌症进化事件限制在少数驱动上,而在晚期发展过程中呈现更多元的驱动进化。
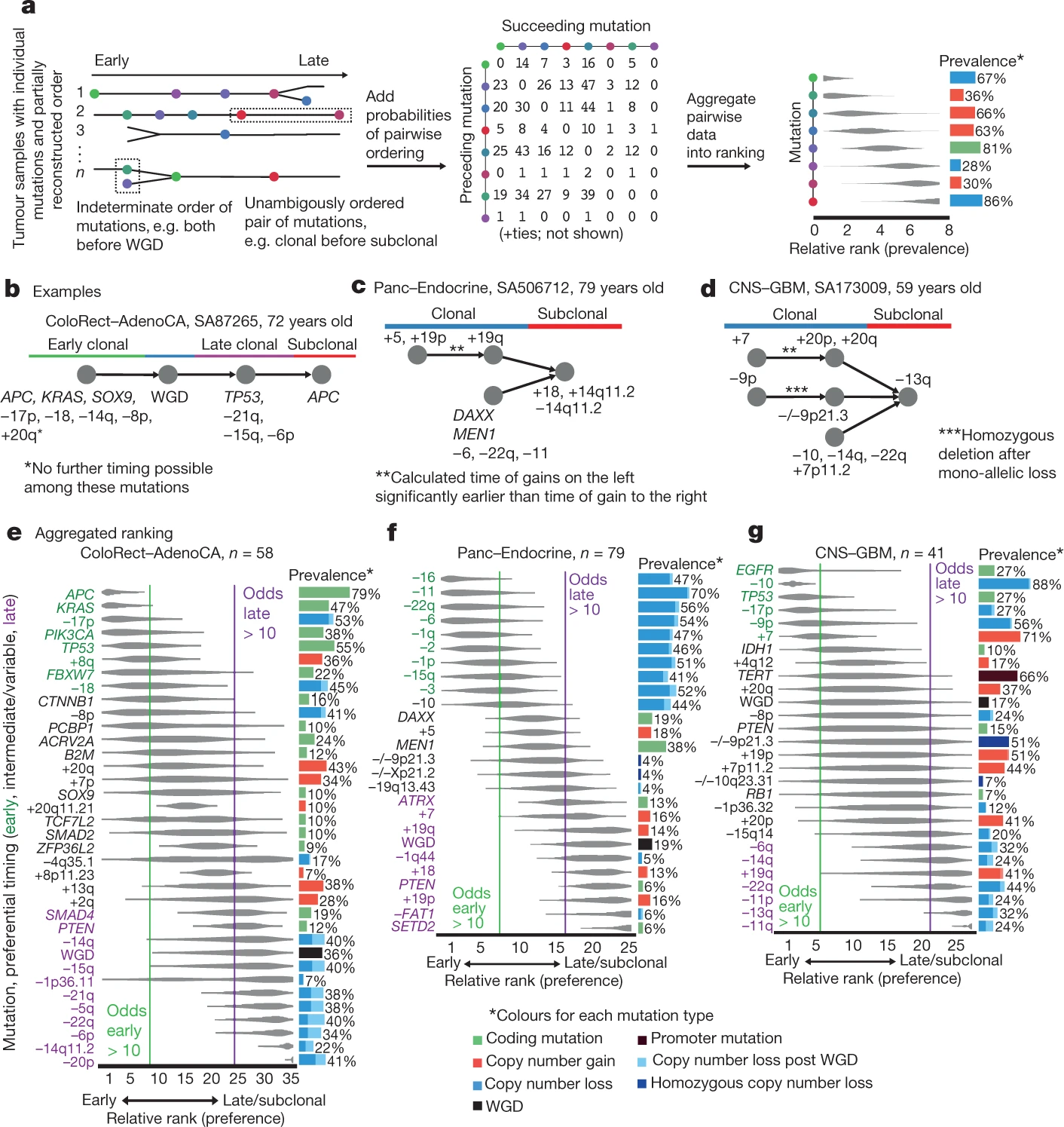
四、体细胞驱动进化事件的相对时间
- 跨样本的概率排序病变分析,检验每个突变在肿瘤进化早期和晚期中的偏好。结果发现APC-KRAS-TP53在肿瘤早期依次有最高的突变几率;拷贝数变异在后期发生。
五、突变印记的形成时间
- 突变印记(Mutational Signatures)定义为由不同因素,如外源或内源DNA损伤,或DNA复制偏差诱发肿瘤基因组形成的不同突变组合。通过在不同时期体细胞突变的分布,发现外源因素诱发的突变(尼古丁印记SBS4和紫外暴露印记SBS7)更多发生在肿瘤进化早期,印记SBS9,SBS12随时间呈递减趋势,而APOEEC(SBS2和SBS13)和SBS38从早期克隆到晚期克隆中都在增长。由有缺陷的错配修复导致的突变印记(SBS6,14,15,20,21,26,44)从克隆到亚克隆时期呈增长趋势。
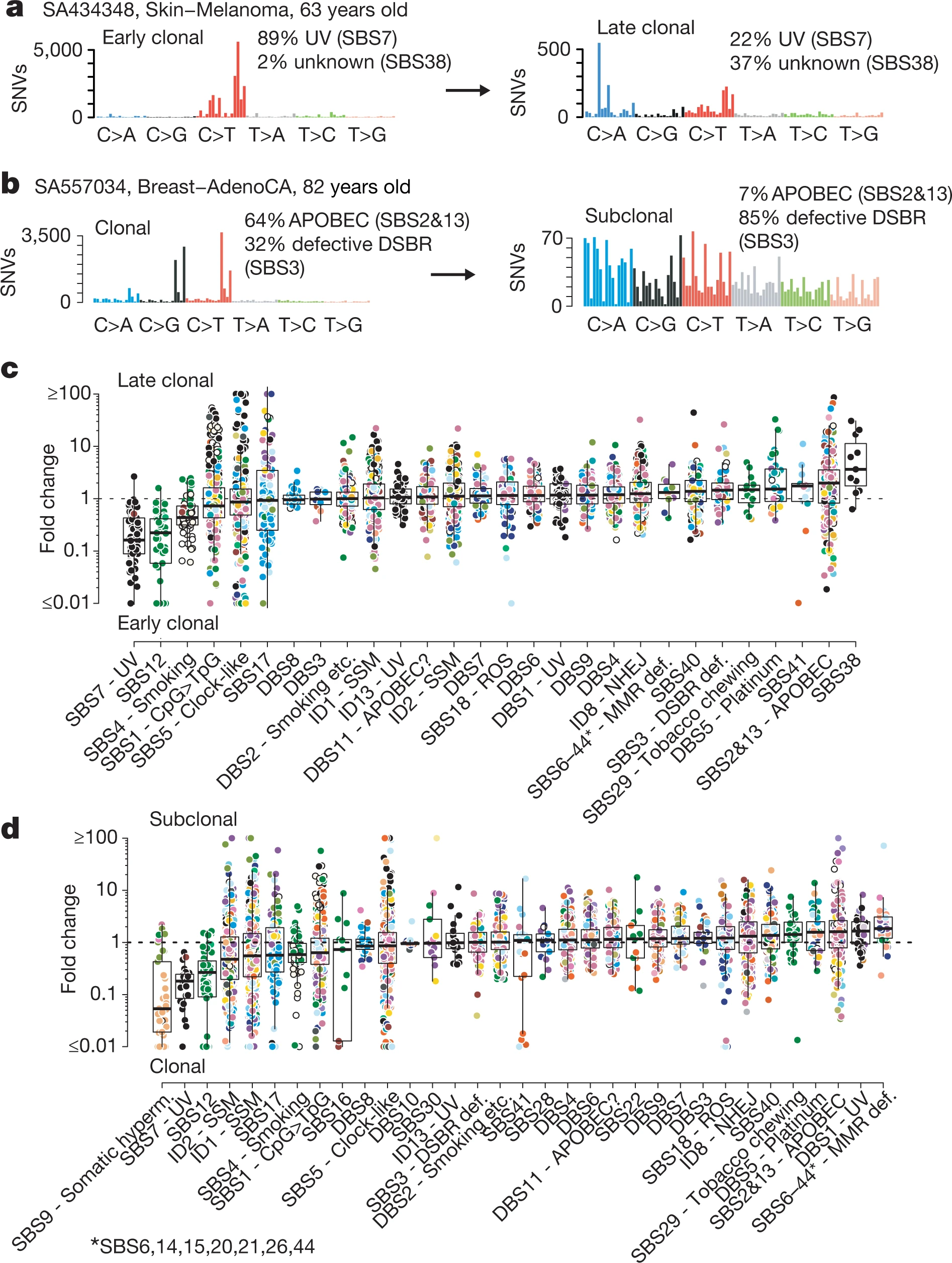
六、时间顺序的肿瘤演化估计
- 分子钟(Molecular Clock)模型结合癌症诊断时间,估计肿瘤演化的时间发展。
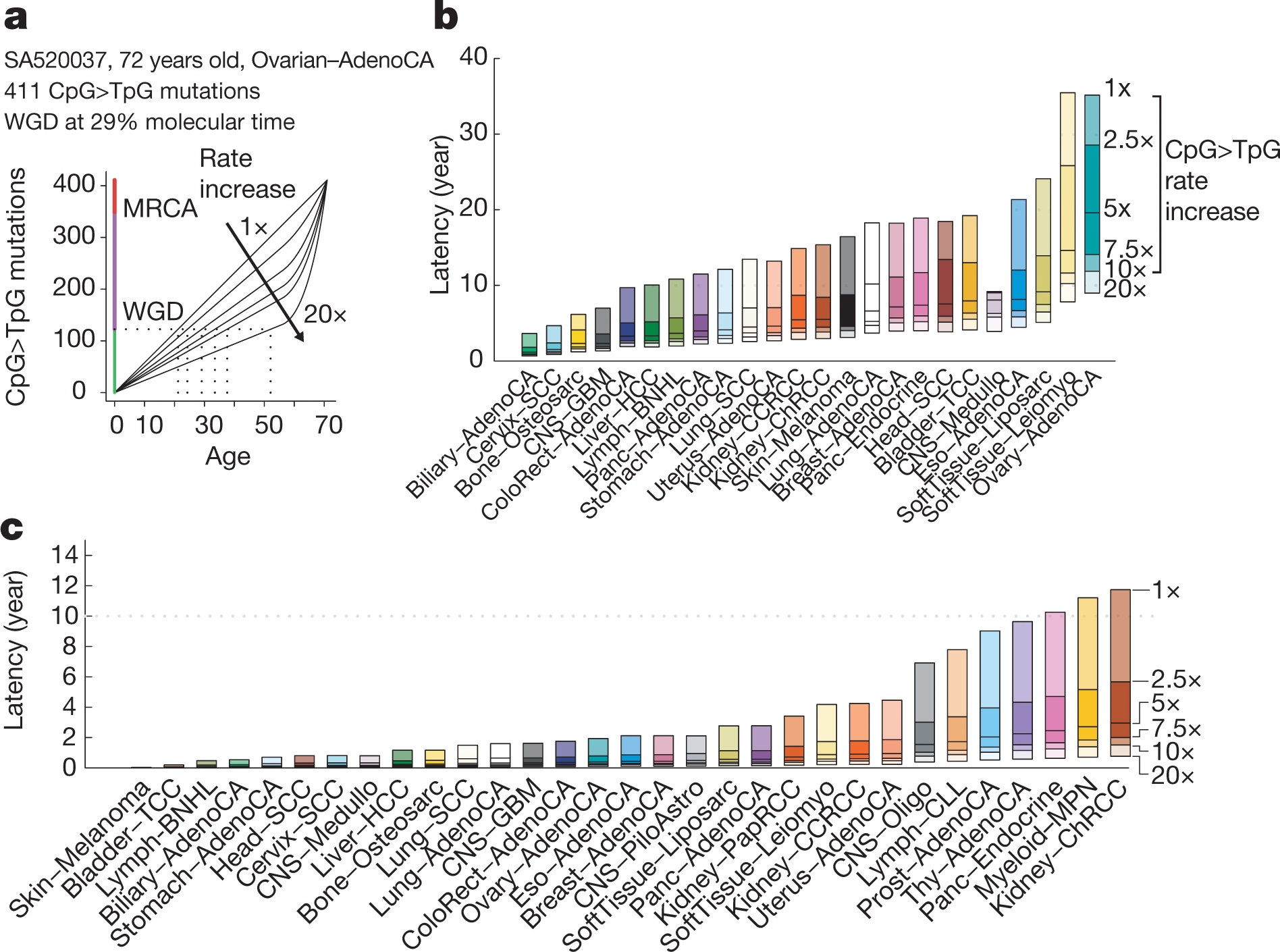
七、癌症发展的一般时间线
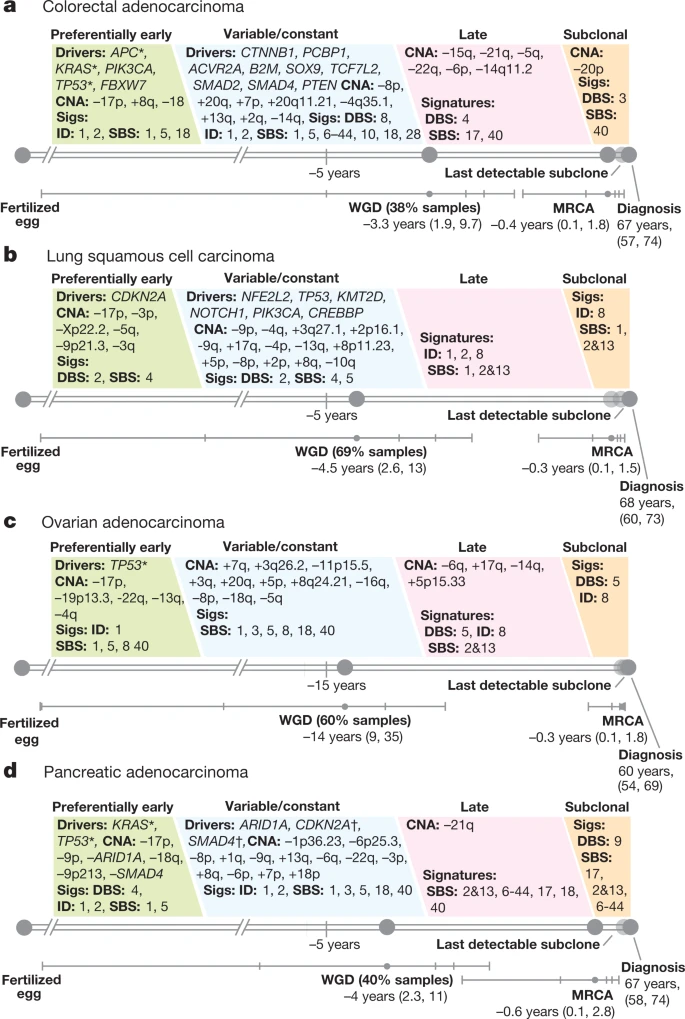
八、讨论