Permutation test
Permutation test又称randomization test,是Fisher[2]和 Pitman[3]等人于20世纪30年代提出的一种统计推断的方法,其属于一种非参数检验,对样本总体分布情况无要求,特别适用于总体分布未知的小样本数据,即使样本数据小到无法使用比如说t检验。Permutation test也可以用于分布未知的大样本量的数据,因此,其应用非常广泛。
基本原理:
Permutation test一般通过对两组样本进行顺序上的随机置换,并重新计算统计检验量,把上述过程重复多遍(比如说1000遍),就可以构造出统计检验量的经验分布,然后对比两组样本的统计检验量和构造出的统计检验量经验分布,就可以计算求出P值。
由于早期受到计算机技术的限制(因为Permutation test要产生大量的随机样本组合),Permutation test只能用于小样本数据的检验,现在随着目前计算机技术的发展,可以在短时间内产生大量的随机样本组合,因此目前Permutation test可以用于样本量较大的数据。
实例分析:
欲研究某新的教学方法能否显著提高学生的数学成绩。两组被试,每组10个人,A组采用传统教学方法,而B组采用这种新的教学方法,经过一个学期的教学后进行测试,并统计两组被试的成绩,如下所示:
A组成绩:65,75,43,80,67,68,54,78,80,62
B组成绩:91,69,73,87,75,71,89,64,70,95
针对这个例子,采用Permutationtest的步骤如下:
第一步:建立H0假设,即新不会提高学生的成绩,或者说新、旧方法并无差别;
第二步:计算统计检验量,这里同样计算两组被试的均值之差,Ms=-13.2;
第三步:把A组和B组成绩进性混合,
AB:65,75,43,80,67,68,54,78,80,62,91,69,73,87,75,71,89,64,70,95
从AB中随机抽取10作为新的A组(计为A1组)成绩,剩下的作为B组成绩(计为B1组),并重新计算统计检验量,计为Mn;
上述随机置换步骤重复若干次(如1000次)可以得到Mn的经验分布;
第四步:计算Mn中大于Ms的个数(计为n),那么P=n/随即置换次数。
作者[1]用Matlab编程,实现上述Permutation test,统计量Mn的经验分布如下图所示:
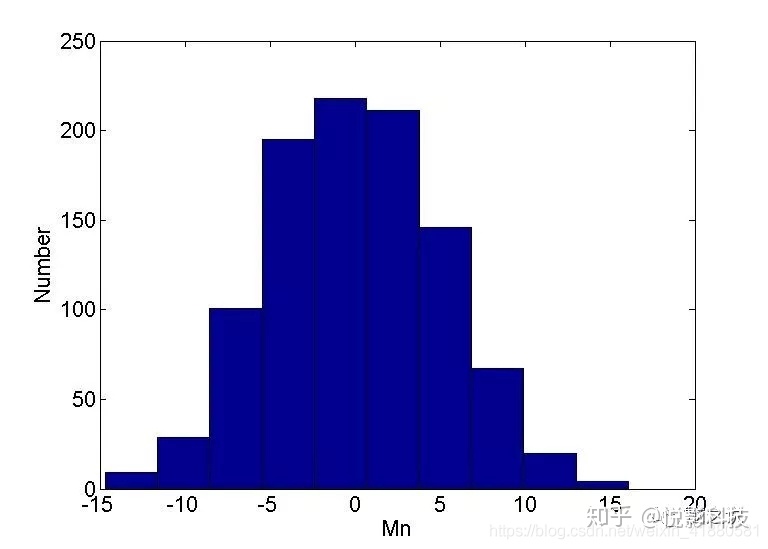
得到的P值为:0.0090,说明新的教学方法可以显著提高学生的成绩。
RegioneR 中关于permutation test几个函数的原理
作为一个生物狗,作者本人想谈谈一个用permutation产生随机背景的方式来进行显著性检验的软件RegioneR所涉及到的相关的统计原理。
RegioneR主要被用来进行关于基因组的Region Sets(比如GC island, 启动子,增强子等)之间或者Region Sets和表观基因组信号区域(H3K27ac,H3K4me3等)之间关联的显著性检验。
可以解决的生物学问题有以下:
- Do my regions contain more SNPs than expected by chance? (该区域内相比于随机背景是否显著富集更多的SNP位点)
- Are my regions enriched in repetitive regions?(该区域是否在重复序列区域富集)
- Are my regions significantly closer to genes?(该区域是否显著邻近于基因区域)
- Are DNA methylation levels in my Region Sets higher than expected?(该区域内的DNA甲基化水平是否显著高于随机背景)
- Do my regions overlap with recurrent SCNAs(Somatic copy number alterations) in my samples?(该区域内是否体细胞拷贝数变异区域显著重叠)
- Are my ChIP peaks associated with that other histone mark?(ChIP-seq peak是否显著和组蛋白表观修饰标记相关)
- Do my ChIP regions associate with repressed genes?(该ChIP-seq区域是否显著和被抑制转录的基因相关)
随机化策略
special <- toGRanges(system.file("extdata", "my.special.genes.txt", package="regioneR"))
all.genes <- toGRanges(system.file("extdata", "all.genes.txt", package="regioneR"))
altered <- toGRanges(system.file("extdata", "my.altered.regions.txt", package="regioneR"))
length(special)
[1] 200
length(all.genes)
[1] 49646
length(altered)
[1] 8
numOverlaps(special, altered)
[1] 114
通过对一个特定的基因集区域和发生拷贝数增加的区域进行Overlap的数目计算(这里计算的是与拷贝数增加的区域的特定基因集区域的数目),发生了114次重叠。这个数目看上去挺多的,那么它真的是否算多呢?多少overlap数目算多?随机从基因组中抽出同样多的同样长度分布的区域,和该基因集区域进行Overlap计算重叠数目的水平,和114这个观测到的重叠数目相比,是高还是低,即这个观测重叠数,是随机产生的,还是显著的具有统计学意义的?因此需要采用一些随机化策略生成随机背景,来评估该观测的显著性水平。
References:
[1] https://zhuanlan.zhihu.com/p/328940140
[2]Fisher, R. A., 1935. The Design of Experiments. Oliver & Boyd, Edinburgh.
[3]Pitman, E., 1937. Significance tests which may be applied to samples from any populations: I. Journal of the Royal Statistical Society, B, 4, 119-130